Taking some of the guesswork out of drug discovery
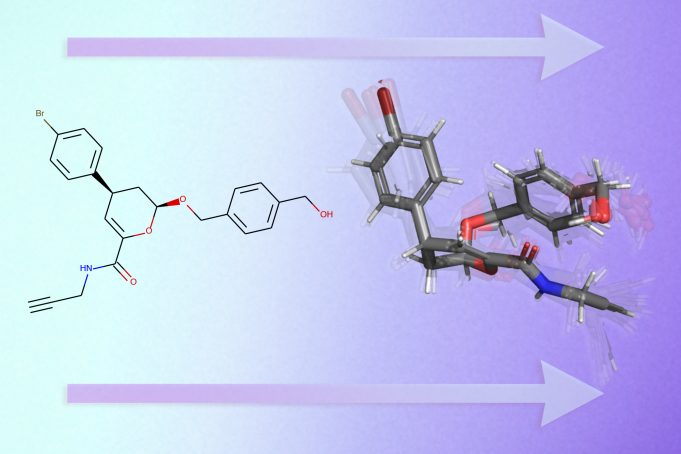
A deep learning model rapidly predicts the 3D shapes of drug-like molecules, which could accelerate the process of discovering new medicines.
In their quest to discover effective new medicines, scientists search for drug-like molecules that can attach to disease-causing proteins and change their functionality. It is crucial that they know the 3D shape of a molecule to understand how it will attach to specific surfaces of the protein.
But a single molecule can fold in thousands of different ways, so solving that puzzle experimentally is a time consuming and expensive process akin to searching for a needle in a molecular haystack.
MIT researchers are using machine learning to streamline this complex task. They have created a deep learning model that predicts the 3D shapes of a molecule solely based on a graph in 2D of its molecular structure. Molecules are typically represented as small graphs.
Their system, GeoMol, processes molecules in only seconds and performs better than other machine learning models, including some commercial methods. GeoMol could help pharmaceutical companies accelerate the drug discovery process by narrowing down the number of molecules they need to test in lab experiments, says Octavian-Eugen Ganea, a postdoc in the Computer Science and Artificial Intelligence Laboratory (CSAIL) and co-lead author of the paper.
“When you are thinking about how these structures move in 3D space, there are really only certain parts of the molecule that are actually flexible, these rotatable bonds. One of the key innovations of our work is that we think about modeling the conformational flexibility like a chemical engineer would. It is really about trying to predict the potential distribution of rotatable bonds in the structure,” says Lagnajit Pattanaik, a graduate student in the Department of Chemical Engineering and co-lead author of the paper.
Other authors include Connor W. Coley, the Henri Slezynger Career Development Assistant Professor of Chemical Engineering; Regina Barzilay, the School of Engineering Distinguished Professor for AI and Health in CSAIL; Klavs F. Jensen, the Warren K. Lewis Professor of Chemical Engineering; William H. Green, the Hoyt C. Hottel Professor in Chemical Engineering; and senior author Tommi S. Jaakkola, the Thomas Siebel Professor of Electrical Engineering in CSAIL and a member of the Institute for Data, Systems, and Society. The research will be presented this week at the Conference on Neural Information Processing Systems.
Mapping a molecule
In a molecular graph, a molecule’s individual atoms are represented as nodes and the chemical bonds that connect them are edges.
GeoMol leverages a recent tool in deep learning called a message passing neural network, which is specifically designed to operate on graphs. The researchers adapted a message passing neural network to predict specific elements of molecular geometry.
Given a molecular graph, GeoMol initially predicts the lengths of the chemical bonds between atoms and the angles of those individual bonds. The way the atoms are arranged and connected determines which bonds can rotate.
GeoMol then predicts the structure of each atom’s local neighborhood individually and assembles neighboring pairs of rotatable bonds by computing the torsion angles and then aligning them. A torsion angle determines the motion of three segments that are connected, in this case, three chemical bonds that connect four atoms.
“Here, the rotatable bonds can take a huge range of possible values. So, the use of these message passing neural networks allows us to capture a lot of the local and global environments that influences that prediction. The rotatable bond can take multiple values, and we want our prediction to be able to reflect that underlying distribution,” Pattanaik says.
Overcoming existing hurdles
One major challenge to predicting the 3D structure of molecules is to model chirality. A chiral molecule can’t be superimposed on its mirror image, like a pair of hands (no matter how you rotate your hands, there is no way their features exactly line up). If a molecule is chiral, its mirror image won’t interact with the environment in the same way.
This could cause medicines to interact with proteins incorrectly, which could result in dangerous side effects. Current machine learning methods often involve a long, complex optimization process to ensure chirality is correctly identified, Ganea says.
Because GeoMol determines the 3D structure of each bond individually, it explicitly defines chirality during the prediction process, eliminating the need for optimization after-the-fact.
After performing these predictions, GeoMol outputs a set of likely 3D structures for the molecule.
“What we can do now is take our model and connect it end-to-end with a model that predicts this attachment to specific protein surfaces. Our model is not a separate pipeline. It is very easy to integrate with other deep learning models,” Ganea says.
A “super-fast” model
The researchers tested their model using a dataset of molecules and the likely 3D shapes they could take, which was developed by Rafael Gomez-Bombarelli, the Jeffrey Cheah Career Development Chair in Engineering, and graduate student Simon Axelrod.
They evaluated how many of these likely 3D structures their model was able to capture, in comparison to machine learning models and other methods.
In nearly all instances, GeoMol outperformed the other models on all tested metrics.
“We found that our model is super-fast, which was really exciting to see. And importantly, as you add more rotatable bonds, you expect these algorithms to slow down significantly. But we didn’t really see that. The speed scales nicely with the number of rotatable bonds, which is promising for using these types of models down the line, especially for applications where you are trying to quickly predict the 3D structures inside these proteins,” Pattanaik says.
In the future, the researchers hope to apply GeoMol to the area of high-throughput virtual screening, using the model to determine small molecule structures that would interact with a specific protein. They also want to keep refining GeoMol with additional training data so it can more effectively predict the structure of long molecules with many flexible bonds.
“Conformational analysis is a key component of numerous tasks in computer-aided drug design, and an important component in advancing machine learning approaches in drug discovery,” says Pat Walters, senior vice president of computation at Relay Therapeutics, who was not involved in this research. “I’m excited by continuing advances in the field and thank MIT for contributing to broader learnings in this area.”
This research was funded by the Machine Learning for Pharmaceutical Discovery and Synthesis consortium.
Related Stories


